Methodology
This page explains how the calculator derives carrier frequencies from allele frequencies and computes recurrence risks. All calculations follow Hardy-Weinberg equilibrium principles applied to gnomAD population data.
For Research Use Only
The calculations described here are for research and educational purposes. Results are not validated for clinical diagnostics and must be independently verified by qualified professionals.
Hardy-Weinberg Equilibrium
In a large, randomly mating population, allele and genotype frequencies remain constant across generations. This principle lets us predict how common carrier status is from the frequency of the disease-causing allele alone - without needing direct carrier counts.
For a two-allele system with disease allele frequency q and normal allele frequency p (where p + q = 1), the genotype frequencies are:
| Genotype | Frequency | Meaning |
|---|---|---|
| AA (homozygous normal) | p² | Unaffected, no pathogenic variants |
| Aa (heterozygous carrier) | 2pq | Carries one pathogenic variant |
| aa (homozygous affected) | q² | Affected with two pathogenic variants |
Where q = disease allele frequency, p = 1 − q.
Calculating Carrier Frequency
Allele Frequency Aggregation
Many autosomal recessive conditions are caused by multiple different pathogenic variants in the same gene. The calculator handles this by summing allele frequencies across all qualifying variants:
- For each variant:
variant_AF = (exome_AC + genome_AC) / (exome_AN + genome_AN) - Combined disease allele frequency:
q = Σ(variant_AF)- the sum of all individual variant allele frequencies
This approach is important for genes like CFTR, where each individual pathogenic variant is rare but the combined carrier frequency is clinically significant (approximately 1 in 25 for European populations).
From Allele Frequency to Carrier Frequency
The carrier frequency is the heterozygous genotype frequency 2pq. For rare disease alleles (small q), p ≈ 1, so:
carrier_frequency = 2pq ≈ 2q
The calculator uses this approximation: carrier_frequency = 2 × q
Why the Approximation Works
For a gene with combined allele frequency q = 0.02 (2%): exact 2pq = 2 × 0.98 × 0.02 = 0.0392. Approximation 2q = 0.04. The difference is 0.0008 - negligible for clinical purposes. The approximation is most accurate when q is small; for virtually all autosomal recessive conditions (q < 0.05), the error remains under 1%.
Population-Specific Calculations
gnomAD provides separate allele counts (AC) and allele numbers (AN) for each population. The calculator uses these to compute population-specific carrier frequencies:
- Each population has its own
q_pop = Σ(AC_pop / AN_pop)andcarrier_freq_pop = 2 × q_pop - Population-specific values are displayed alongside the global figure
- A founder effect is flagged when a population's carrier frequency exceeds 5× the global carrier frequency
This population breakdown is clinically important: carrier frequencies can vary substantially between populations. For example, the CFTR carrier frequency in Ashkenazi Jewish individuals is markedly higher than the global average.
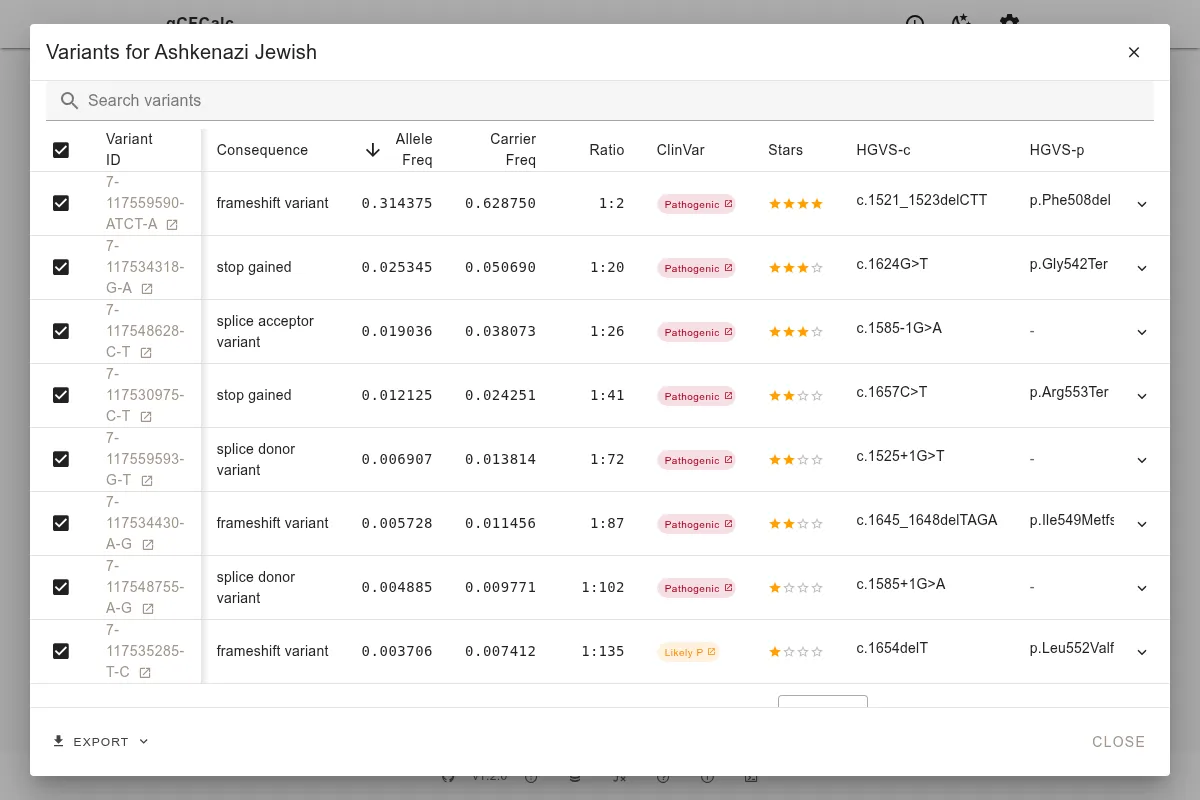
Recurrence Risk
The recurrence risk for offspring depends on the index individual's genetic status. The calculator uses two different formulas based on whether the index individual is a carrier or is affected.
Heterozygous Carrier (risk = carrier_frequency / 4)
For a known carrier, the recurrence risk for offspring requires both parents to pass on a pathogenic allele:
- Partner's carrier probability ≈ population carrier frequency (2q)
- Probability that both carrier parents pass on the pathogenic allele = 1/4
Risk = partner_carrier_prob × 1/4 = carrier_frequency / 4
This status applies when the index individual has one confirmed pathogenic variant.
Affected Individual (risk = carrier_frequency / 2)
For an affected individual (homozygous, compound heterozygous confirmed, or compound heterozygous assumed), one parent is an obligate carrier. The recurrence risk changes:
- Partner's carrier probability ≈ population carrier frequency (2q)
- Probability that the carrier partner passes on the pathogenic allele = 1/2 (only one generation of carrier uncertainty)
Risk = partner_carrier_prob × 1/2 = carrier_frequency / 2
Important Distinction
The risk divisor changes based on status: ÷4 for heterozygous carriers, ÷2 for affected individuals (homozygous, compound het confirmed, or compound het assumed). This reflects whether the index individual's own pathogenic allele status is inferred (carrier of one) or confirmed from both alleles (affected).
Assumptions and Limitations
- Hardy-Weinberg assumes random mating - this holds well for rare recessive conditions where selection pressure is minimal
- gnomAD is a population reference, not a diagnostic database - some ancestries are underrepresented and population-specific estimates may have wide confidence intervals
- The approximation 2pq ≈ 2q introduces less than 0.1% error for allele frequencies under 5%
- Allele frequency aggregation assumes independent pathogenic variants - no linkage disequilibrium is modeled between variants in the same gene
- The calculator does not account for reduced penetrance or variable expressivity - conditions where penetrance is incomplete require additional expert assessment
See Data Sources for details on gnomAD version selection and population coverage. See Filters for how qualifying variants are selected before allele frequencies are summed.
Calculate Now
Apply these methods with real gnomAD data: