Getting Started
This page walks you through a complete calculation from start to finish. You will search for a gene, set the individual's status, choose a frequency source, and review results - all in under a minute.
For Research Use Only
This tool is intended for research and educational purposes. It is not a validated clinical diagnostic tool. Any outputs must be independently verified by qualified professionals before use in a clinical context.
Before You Begin
Open the calculator in a new tab: Launch Calculator. No account or installation required - the calculator runs entirely in your browser.
Step 1: Search for a Gene
The first step is to select the gene you want to analyze. The search field accepts standard gene symbols such as CFTR, HFE, or BRCA1. Start typing and the autocomplete field will query gnomAD in real time, showing matching genes as you type. Select your gene from the list to continue.
Before searching, you can choose which gnomAD version to query using the version selector above the search field. The default is v4.1 (GRCh38), which is the current recommended version with the largest dataset. v2.1.1 (GRCh37) is available for legacy comparisons using the older reference genome. v3.1.2 (GRCh38) provides genome-only data and includes the Amish population, which is useful for specific founder effect analyses. See Data Sources for a detailed comparison of the versions.
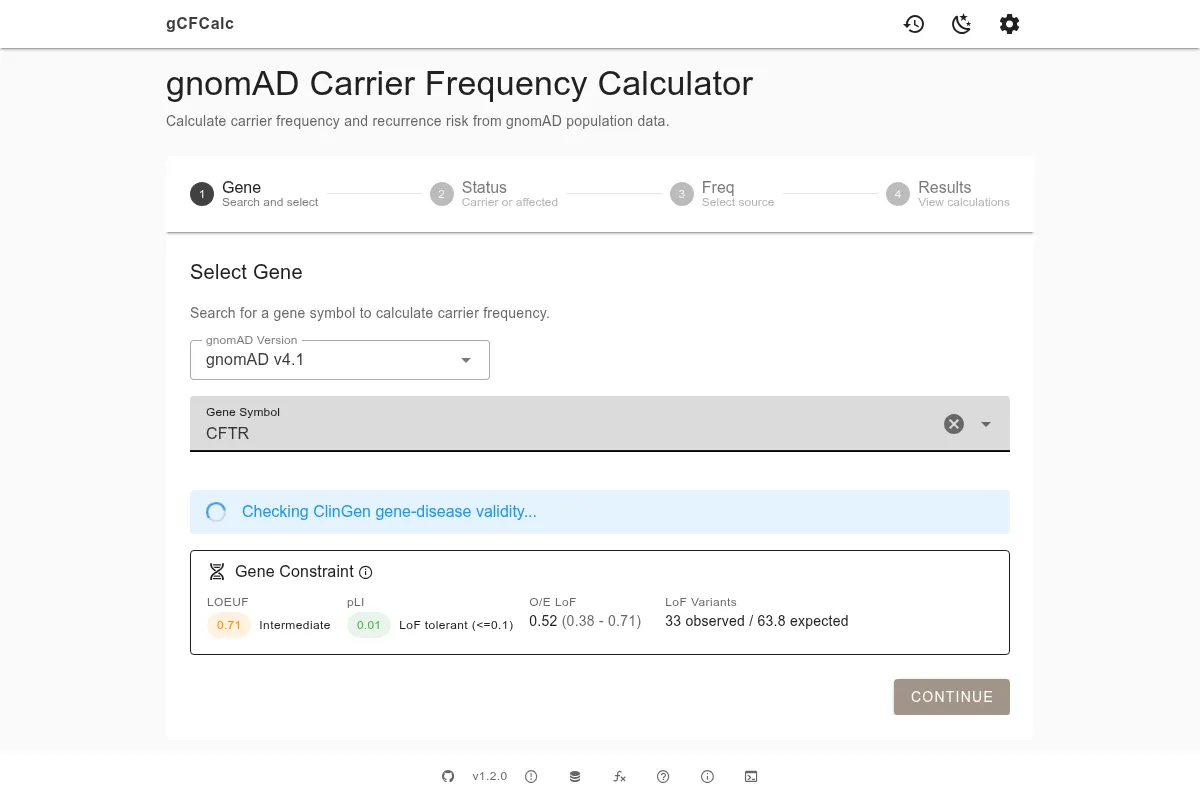
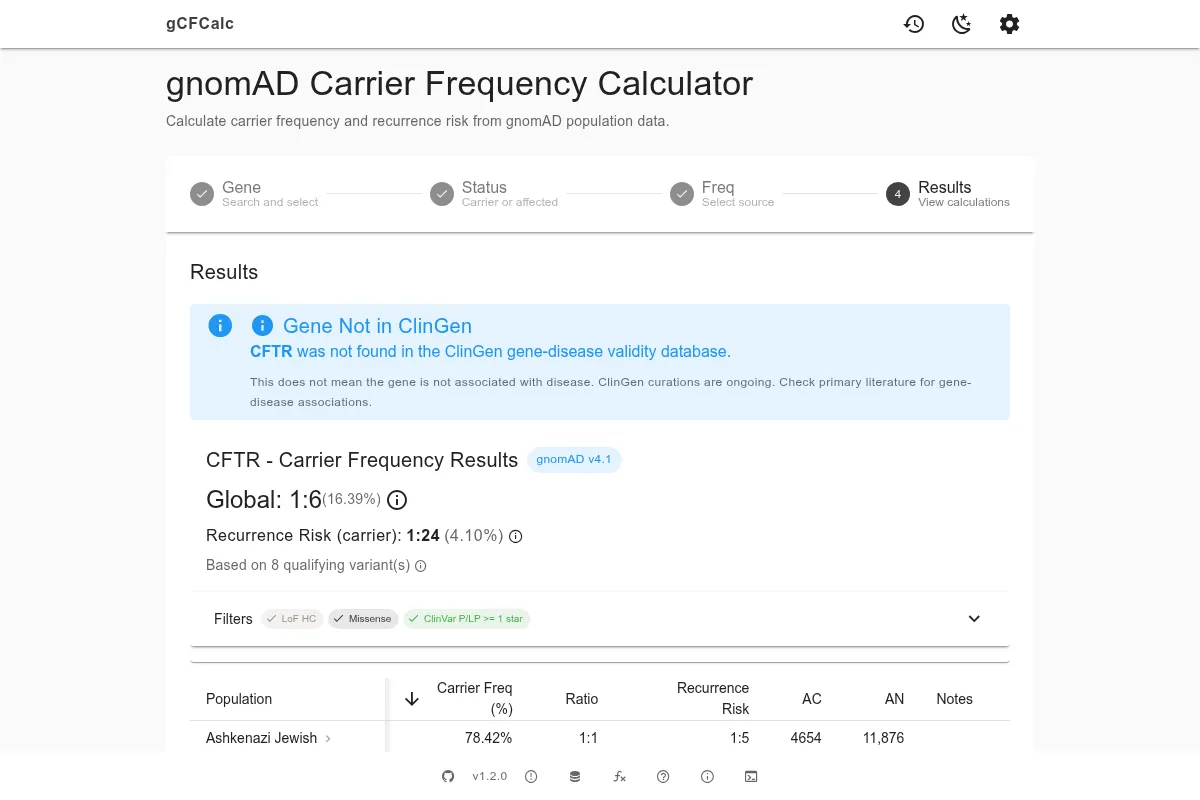
After selecting a gene, the calculator displays a gene constraint panel showing three scores: pLI (probability of loss-of-function intolerance), LOEUF (loss-of-function observed/expected upper bound fraction), and mis_z (missense z-score). High pLI (close to 1) and low LOEUF (below 0.35) indicate that the gene is intolerant of loss-of-function variants - which helps you assess whether the variants found are likely to be clinically significant. If ClinGen gene-disease validity data is available for the gene, a banner will appear showing the validity classification (e.g., Definitive, Strong, Moderate). This information is advisory only and does not affect which variants the calculator includes.
Step 2: Set the Individual's Status
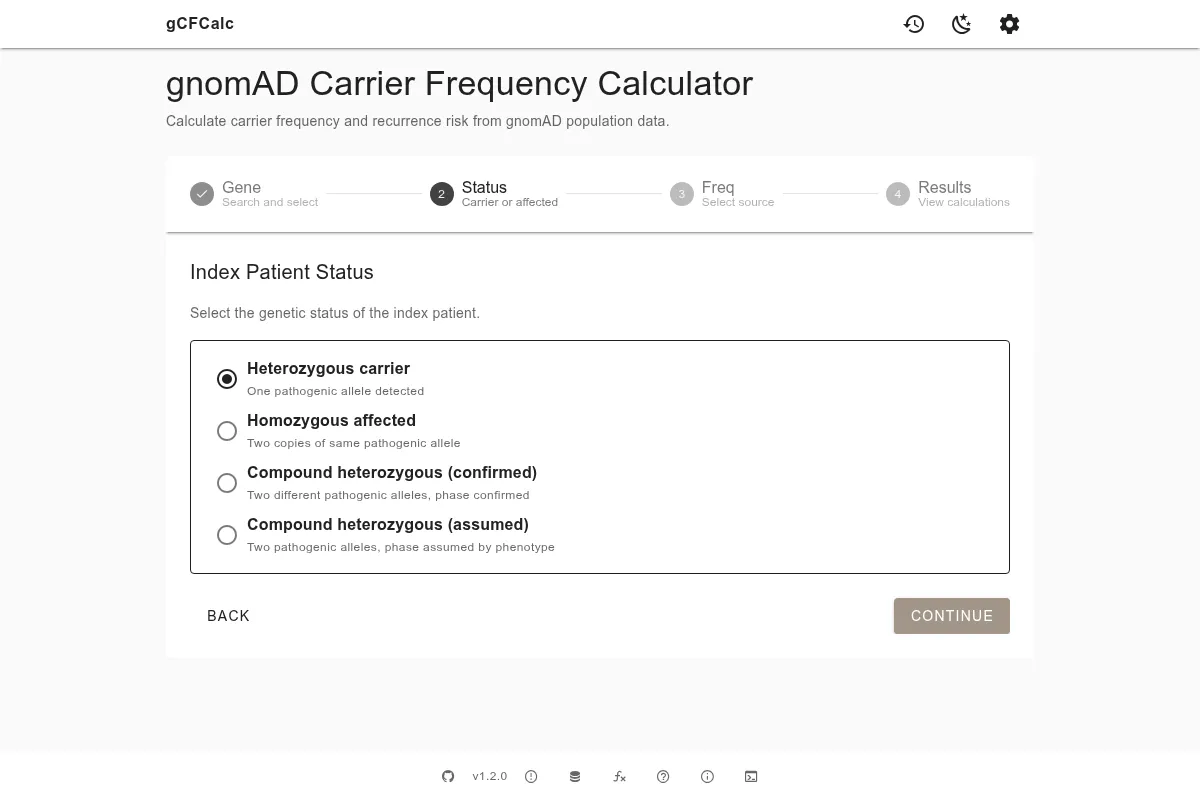
The status describes the index individual's genetic findings. This step determines which recurrence risk formula the calculator uses, so it is important to select the correct option for your scenario.
There are four options:
- Heterozygous carrier - the index individual carries one pathogenic variant on one allele. The recurrence risk depends on whether a second individual also carries a variant, so the risk is calculated as: carrier frequency × 1/4 (the probability that both pass on their pathogenic allele). Expressed differently, the recurrence risk = carrier frequency ÷ 4.
- Homozygous affected - the index individual has two copies of the same pathogenic variant. One allele is already confirmed, so the risk formula uses a divisor of 2: recurrence risk = carrier frequency ÷ 2.
- Compound heterozygous (confirmed) - two different pathogenic variants are confirmed on separate alleles. The recurrence risk is the same as for homozygous affected: carrier frequency ÷ 2.
- Compound heterozygous (assumed) - two different pathogenic variants are detected but the phase (which variant is on which allele) has not been confirmed. The same risk formula applies as for confirmed compound heterozygous, but the generated text notes this assumption.
Why Does Status Matter?
The status determines the recurrence risk formula. For a heterozygous carrier, the risk depends on a second individual also being a carrier (probability = carrier frequency) AND both passing on their pathogenic allele (probability = 1/4). For affected individuals, one allele is already confirmed pathogenic, so the divisor is 1/2 rather than 1/4.
Step 3: Choose a Frequency Source
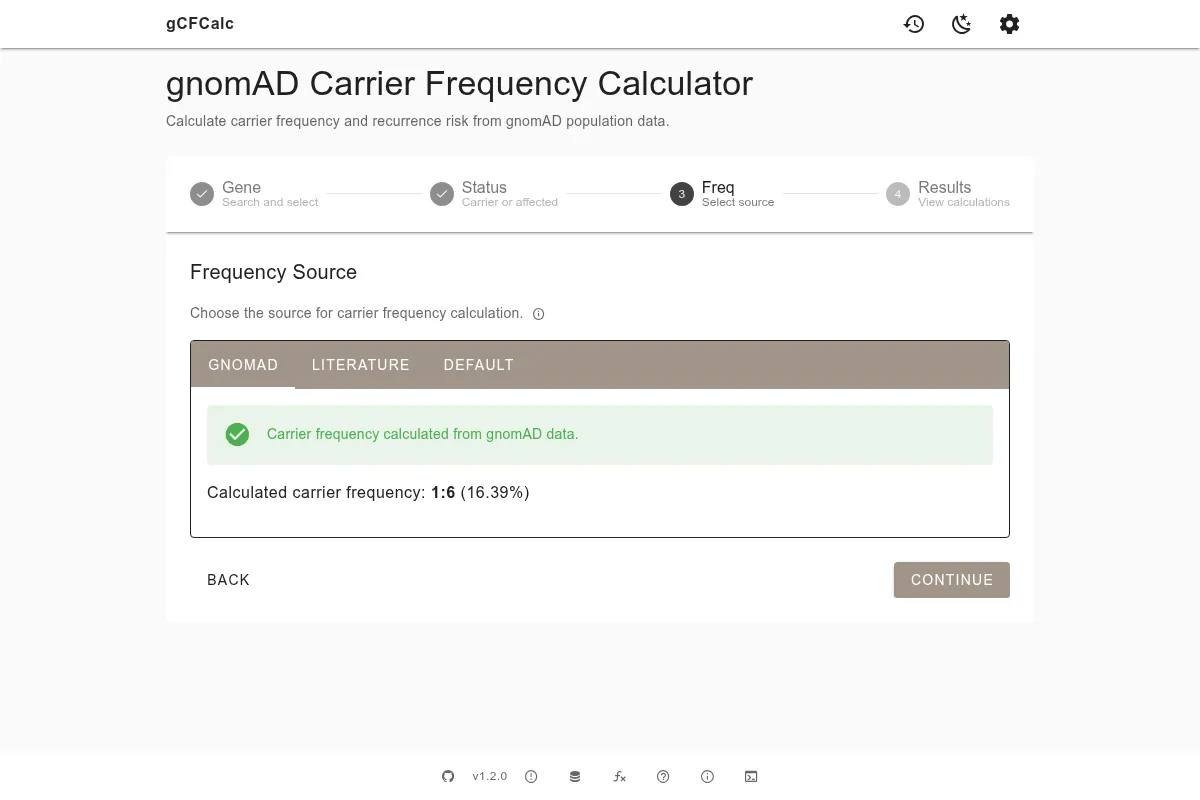
The frequency source step lets you specify where the carrier frequency data comes from. There are three options, presented as tabs:
gnomAD tab (default) - the calculator queries gnomAD directly for all pathogenic variants in your selected gene. A variant table shows each qualifying variant with its allele count, allele number, and allele frequency. Variants are pre-filtered for high-confidence loss-of-function (LoF HC) and ClinVar pathogenic/likely pathogenic entries. You can review the variant list and exclude specific variants if needed - for example, if a variant has disputed pathogenicity. See Filters for a full description of how variants are selected.
Literature tab - enter a carrier frequency from a published study. This is useful when you have a known population-specific value from the literature, such as 1:25 for CFTR in Northern Europeans. You can optionally enter a PubMed ID, which will be cited in the generated text.
Default tab - uses a fallback carrier frequency of 1% (1:100). This is a conservative estimate useful for quick calculations when neither gnomAD data nor literature values are available for a given gene or population.
Step 4: Review Results and Generate Text
The final step shows your calculation results and generates documentation text.
The population table displays carrier frequency and recurrence risk for each gnomAD population. Global values appear first, followed by population-specific breakdowns (African/African-American, Admixed American, Ashkenazi Jewish, East Asian, Finnish, Middle Eastern, Non-Finnish European, South Asian). If one population shows a notably higher carrier frequency than the global average, this may indicate a founder effect - a historically isolated population where a particular variant has become more common. See Methodology for details on the calculation.
The text panel on the right generates documentation based on your selections. You can choose the perspective - affected individual, healthy carrier, or family member - and the language (German or English). For German text, you can select the gender-inclusive style: asterisk (*), colon (😃, or slash (/). The text is automatically populated with your gene, variant, frequency, and risk data.
Use the copy button to copy the text to your clipboard. The share button generates a URL that encodes your full calculation, which you can send to a colleague or save for reference.
If you want to customize which text sections appear or adjust the wording, click the settings icon to open the template editor. See Templates for a full reference of available variables and sections.
Tips
Your searches are saved automatically
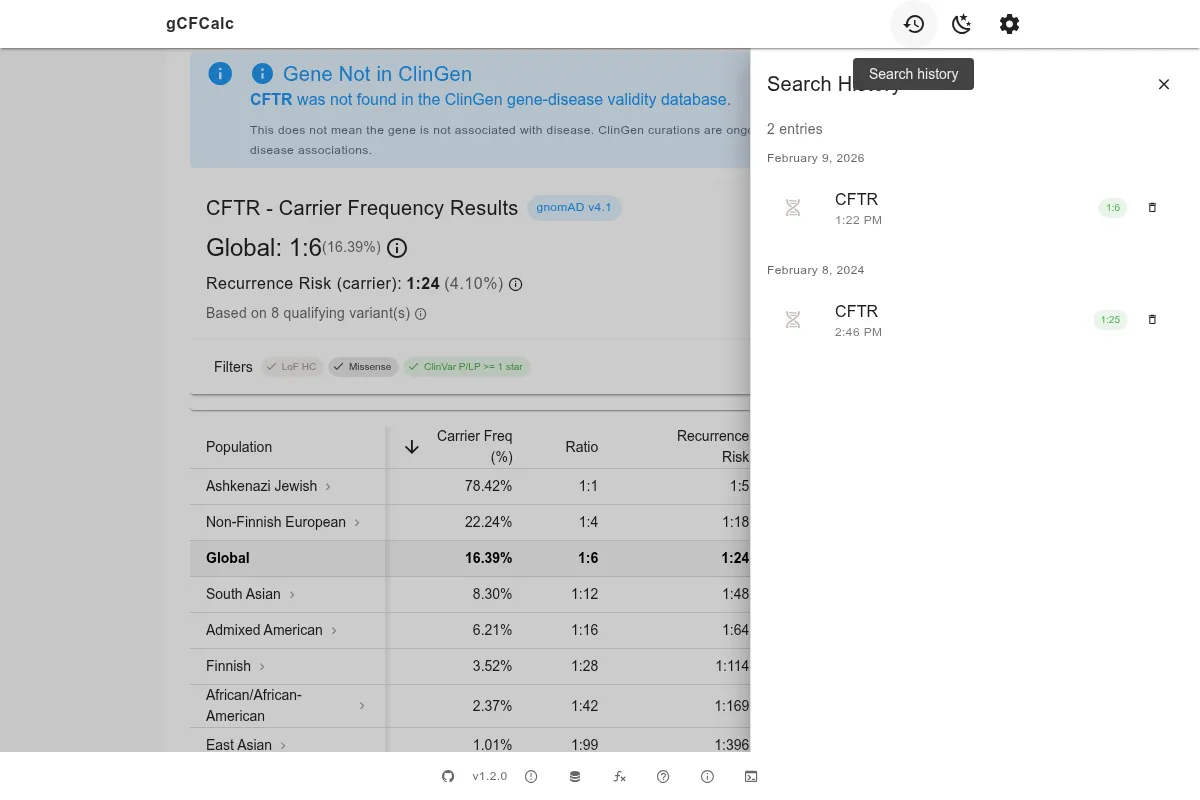
Search History
The calculator saves your recent gene searches locally in your browser. Click the history icon in the top bar to revisit previous calculations without re-entering data.
Works offline
The calculator is a Progressive Web App (PWA). After your first visit, the application is cached in your browser and continues to work without an internet connection. Note that offline mode uses the variant data cached from your last online session, so gnomAD queries require an internet connection when you need fresh data.
What's Next?
- See the Carrier Screening use case for a CFTR scenario with variant exclusion
- See the Text Generation use case for generating documentation with HFE
- Browse the Reference section for methodology, filters, and template details
Start Calculating
You now know the complete workflow. Open the calculator and try it with your gene of interest: